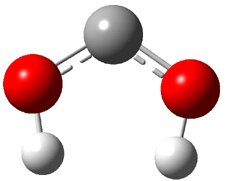
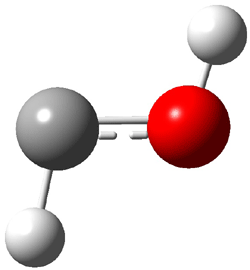
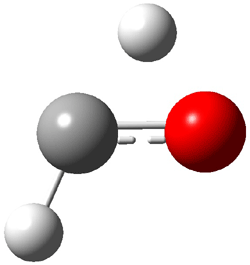
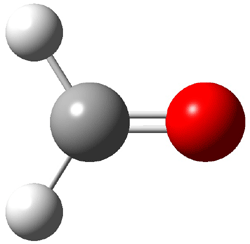
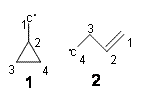Not particularly strong programming at the year’s spring ACS meeting – but one great session in the organic division yesterday. This was the awards session in honor of John Baldwin getting the James Flack Norris Award for physical organic chemistry.
First to speak was James Duncan, who discussed his recent CASSCF computations looking for pseudopericylic [3,3]-sigmatropic migrations. I will be commenting on his latest work in a post that will appear soon.
I had to skip the next talk, but came back to hear John Brauman discuss recent work on the solvation effect in the SN2 reaction. This is an interesting case of where the screening of larger substituents is counterbalanced by geometric changes that lead to greater charge distribution. The net effect is that they cancel each other out, and the methyl,ethyl, iso-propyl, butyl β-effect is negligible.
Next was Peter Schreiner who discussed his carbene work, specifically the enormous tunneling effect observed in hydroxymethylene (see this post). He discussed some new work, that is if anything even more fantastic on methylhydroxycarbene – look for this work perhaps later in 2011.
Last to speak was John Baldwin – and he described his truly tour de force efforts in examining the [1,3]-rearrangements of vinylcyclopropane and vinylcyclobutane. The former work is described in my book, while the later study is still ongoing.
John’s work is amazingly painstaking and careful. I am truly in awe of his dedication in taking on extremely difficult studies that require enormous care. John has really taught us a lot – not just about these rearrangements (they involve diradicals on a flat plateau demanding dynamic analysis – but how to think about a study and then carry it out to fruition so that all details are assessed. A truly deserving recipient!