Capturing buckyballs involves molecular design based on non-covalent interactions. This poses interesting challenges for both the designer and the computational chemist. The curved surface of the buckyball demands a sequestering agent with a complementary curved surface, likely an aromatic curved surface to facilitate π-π stacking interactions. For the computational chemist, weak interactions, like dispersion and π-π stacking demand special attention, particularly density functionals designed to account for these interactions.
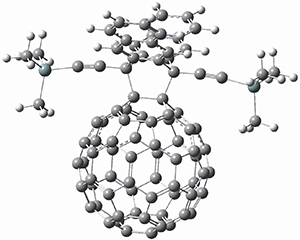
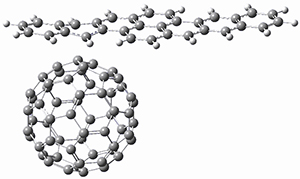
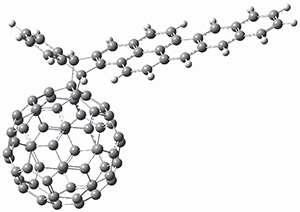
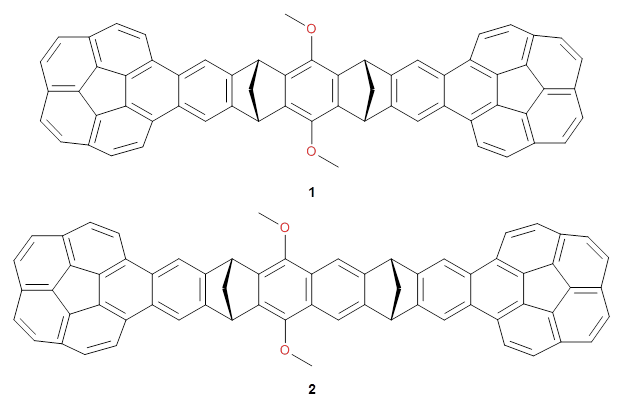
Two very intriguing new buckycatchers were recently prepared in the Sygula lab, and also examined by DFT.1 Compounds 1 and 2 make use of the scaffold developed by Klärner.2 In these two buckycatchers, the tongs are corranulenes, providing a curved aromatic surface to match the C60 and C70 surface. They differ in the length of the connector unit.
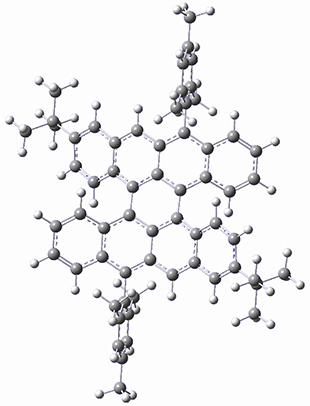
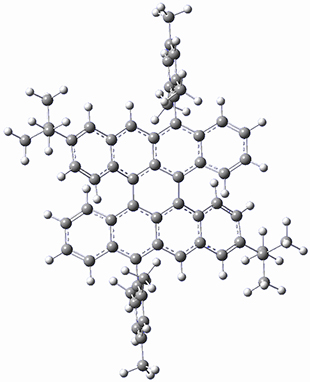
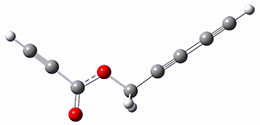
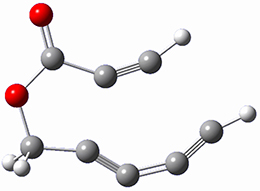
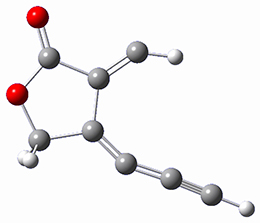
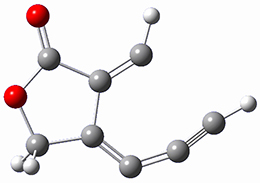
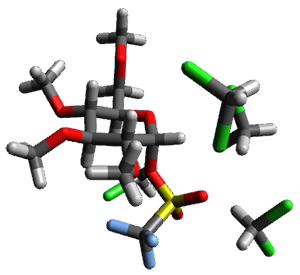
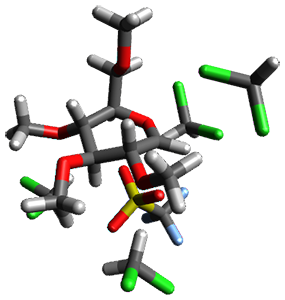
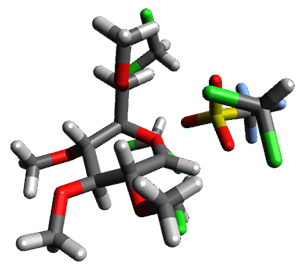
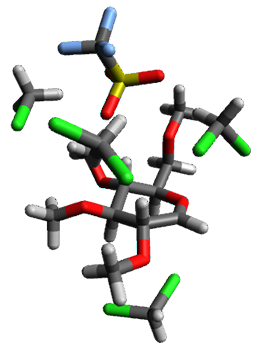
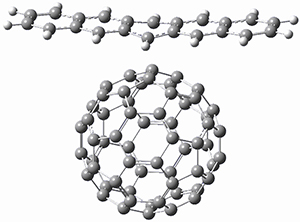
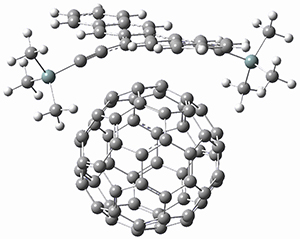
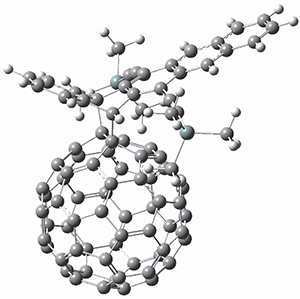
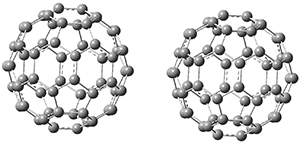
B97-D/TZVP computations of the complex of 1 and 2 with C60 were carried out. The optimized structures are shown in Figure 1. The binding energies (computed at B97-D/QZVP*//B97-D/TZVP) of these two complexes are really quite large. The binding energy for 1:C60 is 33.6 kcal mol-1, comparable to some previous Buckycatchers, but the binding energy of 2:C60 is 50.0 kcal mol-1, larger than any predicted before.
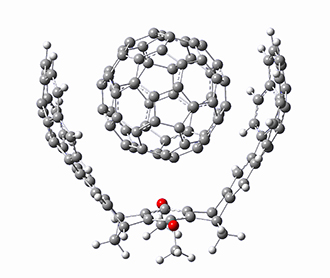
1 |
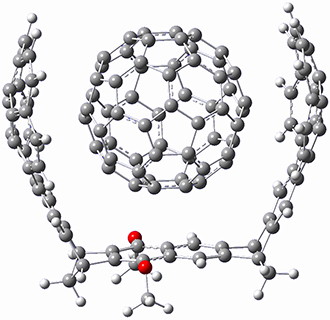
2 |
Figure 1. B97-D/TZVP optimized geometries of 1:C60and 2:C60.
Measurement of the binding energy using NMR was complicated by a competition for one or two molecules of 2 binding to buckyballs. Nonetheless, the experimental data show 2 binds to C60 and C70 more effectively than any previous host. They were also able to obtain a crystal structure of 2:C60.
References
(1) Abeyratne Kuragama, P. L.; Fronczek, F. R.; Sygula, A. "Bis-corannulene Receptors for Fullerenes Based on Klärner’s Tethers: Reaching the Affinity Limits," Org. Lett. 2015, ASAP, DOI: 10.1021/acs.orglett.5b02666.
(2) Klärner, F.-G.; Schrader, T. "Aromatic Interactions by Molecular Tweezers and Clips in Chemical and Biological Systems," Acc. Chem. Res. 2013, 46, 967-978, DOI: 10.1021/ar300061c.
InChIs
1: InChI=1S/C62H34O2/c1-63-61-57-43-23-45(41-21-37-33-17-13-29-9-5-25-3-7-27-11-15-31(35(37)19-39(41)43)53-49(27)47(25)51(29)55(33)53)59(57)62(64-2)60-46-24-44(58(60)61)40-20-36-32-16-12-28-8-4-26-6-10-30-14-18-34(38(36)22-42(40)46)56-52(30)48(26)50(28)54(32)56/h3-22,43-46H,23-24H2,1-2H3/t43-,44+,45+,46-
InChIKey=RLOJCVYXCBOUQB-RYSLUOGPSA-N
2: InChI=1S/C66H36O2/c1-67-65-51-24-45-43-23-44(42-20-38-34-16-12-30-8-4-27-3-7-29-11-15-33(37(38)19-41(42)43)59-55(29)53(27)56(30)60(34)59)46(45)25-52(51)66(68-2)64-50-26-49(63(64)65)47-21-39-35-17-13-31-9-5-28-6-10-32-14-18-36(40(39)22-48(47)50)62-58(32)54(28)57(31)61(35)62/h3-22,24-25,43-44,49-50H,23,26H2,1-2H3/t43-,44+,49+,50-
InChIKey=JAUUHTKCNSNBMD-NETXOKAWSA-N