Use of computed NMR chemical shifts in structure determination is really growing fast. Presented here are a couple of recent examples.
Nguyen and Tantillo used computed chemical shifts with the DP4 analysis to identify the structure of three terpenes 1-3.1 They optimized the geometries of all of the diastereomers of each compound, along with multiple conformations of each diastereomer, at B3LYP/6-31+G(d,p) and then computed the chemical shifts at SMD(CHCl3)–mPW1PW91/6-311+G(2d,p). The chemical shifts were Boltzmann weighted including all conformations within 3 kcal mol-1 of the lowest energy structure.
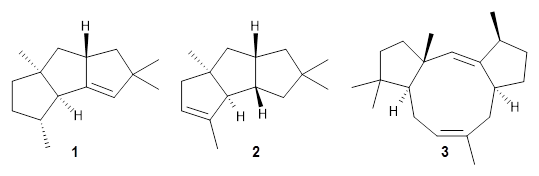
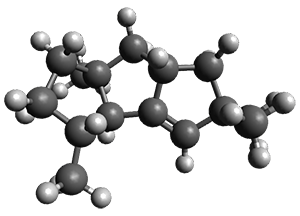
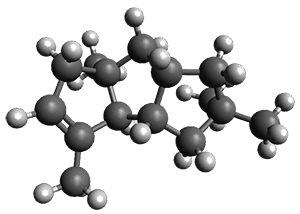
For 1, the DP4 analysis using just the proton shifts predicted a different isomer than using the carbon shifts, but when combined, DP4 predicted the structure, with 98.8% confidence, shown in the scheme above, and in Figure 1. For 2, the combined proton and carbon shift analysis with DP4 indicated a 100% confidence of the structure shown in the scheme and Figure 1. Lastly, for 3, which is more complicated due to the conformations of the 9-member ring, DP4 predicts with 100% confidence the structure shown in the scheme and Figure 1.
1 |
2 |
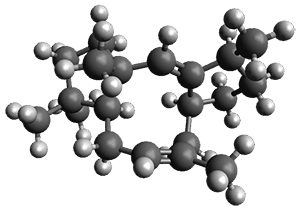
3 |
Figure 1. Optimized geometries of 1-3.
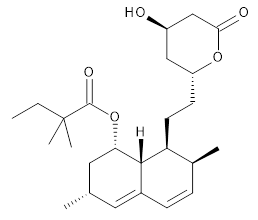
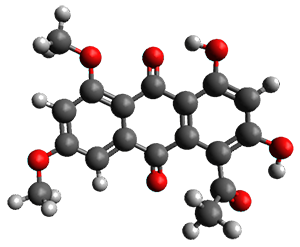
Feng, Davis and coworkers have examined a series of anthroquionones from Australian marine sponges.2 The structure of one compound was a choice of two options: 4 or 5. Initial geometries were obtain by molecular mechanics and the low energy isomers were then reoptimized at B3LYP/6-31+G(d,p). The chemical shifts were computed using PCM/MPW1PW91/6-311+G(2d,p). Application of the DP4 method indicate the structure to be 4 with a 100% confidence level. The lowest energy conformer of 4 is shown in Figure 2.
|
Figure 2. Optimized geometry of 4.
References
1) Nguyen, Q. N. N.; Tantillo, D. J. “Using quantum chemical computations of NMR chemical shifts to assign relative configurations of terpenes from an engineered Streptomyces host,” J. Antibiotics 2016, 69, 534–540, DOI: 10.1038/ja.2016.51.
2) Khokhar, S.; Pierens, G. K.; Hooper, J. N. A.; Ekins, M. G.; Feng, Y.; Rohan A. Davis, R. A. “Rhodocomatulin-Type Anthraquinones from the Australian Marine Invertebrates Clathria hirsuta and Comatula rotalaria,” J. Nat. Prod., 2016, 79, 946–953, DOI: 10.1021/acs.jnatprod.5b01029.
InChIs
1: InChI=1S/C15H24/c1-10-5-6-15(4)8-11-7-14(2,3)9-12(11)13(10)15/h9-11,13H,5-8H2,1-4H3/t10-,11+,13-,15+/m1/s1
InChIKey=KVSCZIPUFBVHBM-OICBVUGWSA-N
2: InChI=1S/C15H24/c1-10-5-6-15(4)8-11-7-14(2,3)9-12(11)13(10)15/h5,11-13H,6-9H2,1-4H3/t11-,12-,13+,15-/m0/s1
InChIKey=ZLYGJLHCPYVGDA-XPCVCDNBSA-N
3: InChI=1S/C20H32/c1-14-6-9-18-19(3,4)10-11-20(18,5)13-17-15(2)7-8-16(17)12-14/h6,13,15-16,18H,7-12H2,1-5H3/b14-6-,17-13-/t15-,16-,18-,20+/m0/s1
InChIKey=JZGOFJIAHJJJDK-ICZJPRMTSA-N
4: InChI=1S/C18H14O7/c1-7(19)13-10(20)6-11(21)15-16(13)17(22)9-4-8(24-2)5-12(25-3)14(9)18(15)23/h4-6,20-21H,1-3H3
InChIKey=MPQMZEXRJVMYBT-UHFFFAOYSA-N
5: InChI=1S/C18H14O7/c1-7(19)13-10(20)6-11(21)15-16(13)14-9(17(22)18(15)23)4-8(24-2)5-12(14)25-3/h4-6,20-21H,1-3H3
InChIKey=WIKIUXNPFURKNF-UHFFFAOYSA-N