Reid, Rathore and colleagues report on the attempted preparation of the interesting molecule 1,3,5-trifluorenylcyclohexane (TFC) 1.1 They had hoped to prepare it by subjecting the precursor 2 to acid, which might then undergo a Friedel-Crafts reaction to prepare the last fluorenyl group, and subsequent loss of a proton would give 1. Unfortunately, they could not get this step to occur, even at high temperature and for long reaction times. What made it particularly frustrating was that they could get 3 to react under these conditions to give 1,4-difluorenylcyclohexane (14-DFC) 4, and convert 5 into 1,4-difluorenylcyclohexane (13-DFC) 6.
|
|
|
|
|
|
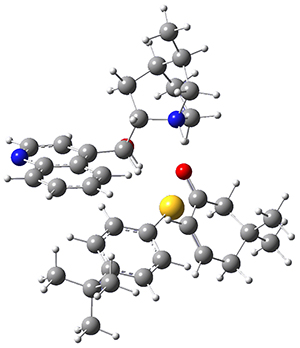
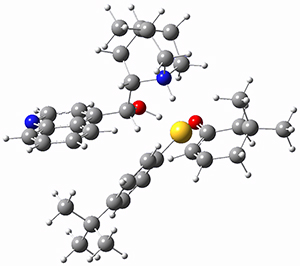
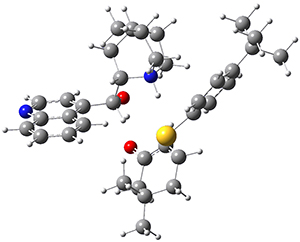
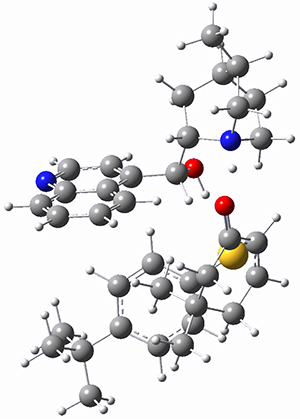
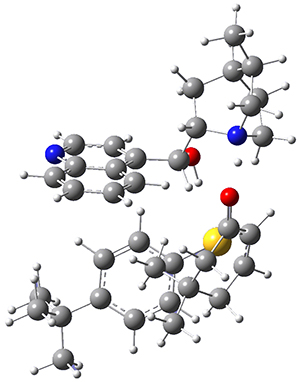
To get at why 1 could not be formed they utilized PCM(CH2Cl2)/M06-2X/6-31G(d) calculations. The lowest energy conformations of 1 and 4 are shown in Figure 1. While 4 is in a chair conformation, 1 is not in a chair conformation since this would bring the three fluorenyl groups into very close contact. Instead, the cyclohexyl ring of 1 adopts a twist-boat conformation, with a much flattened ring. They estimate that 1 is strained by about 17 kcal mol-1, with 10 kcal mol-1 coming from strain in the twist-boat conformation and another 7 kcal mol-1 of strain due to steric crowding of the fluorenyl groups.
They next optimized the structures of the intermediates and transition states on the path taking 2 into 1 and 3 into 4. The transition states of the Friedel-Crafts reaction are the highest points on these paths, and their geometries are shown in Figure 1. The barrier through the TS for the Friedel-Crafts step forming 1 is about 17 kcal mol-1 higher than for the barrier to form 4. This very large increase in activation barrier, due to the strains imposed by that third fluorenyl group, explains the lack of reaction. Furthermore, since the reaction 2 → 1 is 2.0 kcal mol-1 endothermic, at high temperature the reaction is likely to be reversible and favors 2.
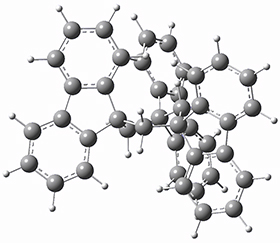
1 |
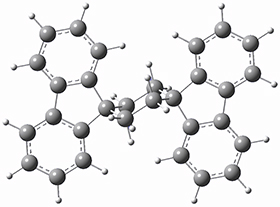
4 |
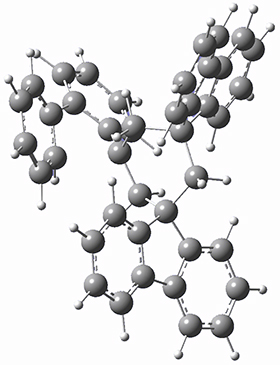
TS to 1 |
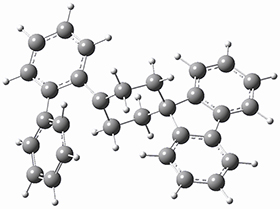
TS to 4 |
Figure 1. PCM(CH2Cl2)/M06-2X/6-31G(d) optimized geometries.
References
(1) Talipov, M. R.; Abdelwahed, S. H.; Thakur, K.; Reid, S. A.; Rathore, R. "From Wires to Cables: Attempted Synthesis of 1,3,5-Trifluorenylcyclohexane as a Platform for Molecular Cables," J. Org. Chem. 2016, DOI: 10.1021/acs.joc.5b02792.
InChIs
1 (TFC): InChI=1S/C42H30/c1-7-19-34-28(13-1)29-14-2-8-20-35(29)40(34)25-41(36-21-9-3-15-30(36)31-16-4-10-22-37(31)41)27-42(26-40)38-23-11-5-17-32(38)33-18-6-12-24-39(33)42/h1-24H,25-27H2
InChIKey=CXXRVQFQMRJLAI-UHFFFAOYSA-N
4 (14-DFC): InChI=1S/C30H24/c1-5-13-25-21(9-1)22-10-2-6-14-26(22)29(25)17-19-30(20-18-29)27-15-7-3-11-23(27)24-12-4-8-16-28(24)30/h1-16H,17-20H2
InChIkey=ZZTDGVHNROVFMK-UHFFFAOYSA-N
6 (13-DFC):InChI=1S/C30H24/c1-2-11-22(12-3-1)24-14-4-5-15-25(24)23-13-10-20-30(21-23)28-18-8-6-16-26(28)27-17-7-9-19-29(27)30/h1-9,11-19H,10,20-21H2
InChIKey=TTZIUDUAWUTKAI-UHFFFAOYSA-N