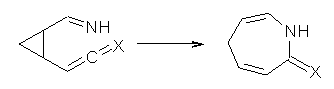
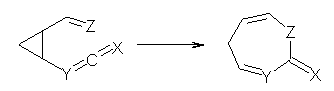
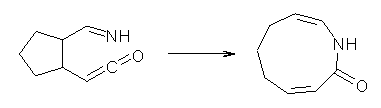
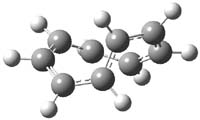
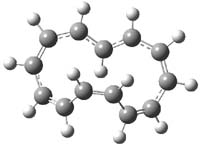
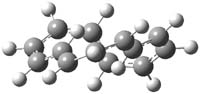
What gives rise to the face selectivity in the epoxidation of the alkene of 1 and 2? And why is the epoxidation of 3 of opposite selectivity? Williams1 argues that the stereoinduction is due to distortional asymmetry, an argument similar to one made recently by Houk2,3 (see this post) and others for cycloaddition reactions.

The major conclusion from this paper is drawn from the potential energy curve that results from out-of-plane bending of the alkenyl hydrogens, as in Figure 1. The bending curves (computed at B3LYP/6-31g(2d,2p)//B3LYP/6-31+G(d))) are asymmetric: bending the hydrogens away from the three-member ring requires less energy than bending them towards the cyclopropyl ring. However, for 3, bending in the two directions is pretty similar, with a slight preference for bending towards the four-member ring.
|
|
|
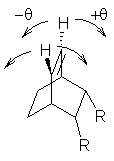
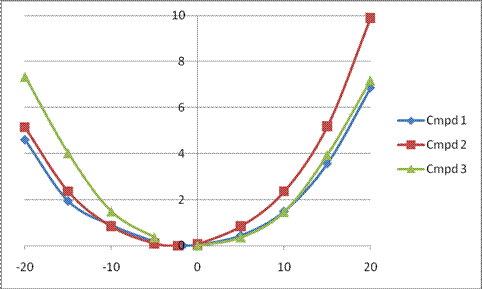
Fig. 1 Energy (kcal mol-1) vs distortion angle of alkenyl hydrogens
This type of bending is part of the distortions that have to occur to reach the transition state, and so Williams argues that the attack from the cyclopropyl face by the oxidant is preferred because of the easier geometric distortion of moving the hydrogen away. Williams makes standard orbital interaction arguments to rationalize the distortion preference.
References
(1) Kolakowski, R. V.; Williams, L. J., "Stereoinduction by distortional asymmetry," Nat. Chem. 2010, 2, 303-307, DOI: 10.1038/nchem.577.
(2)
Xu, L.; Doubleday, C. E.; Houk, K. N., "Dynamics of 1,3-Dipolar Cycloaddition Reactions of Diazonium Betaines to Acetylene and Ethylene: Bending Vibrations Facilitate Reaction," Angew. Chem. Int. Ed. 2009, 48, 2746-2748, DOI: 10.1002/anie.200805906
(3) Xu, L.; Doubleday, C. E.; Houk, K. N., "Dynamics of 1,3-Dipolar Cycloadditions: Energy Partitioning of Reactants and Quantitation of Synchronicity," J. Am. Chem. Soc., 2010, 132, 3029–3037, DOI: http://dx.doi.org/10.1021/ja909372f
InChIs
1: InChI=1/C9H12/c1-2-7-4-3-6(1)8-5-9(7)8/h1-2,6-9H,3-5H2
InChIKey=YNSKHNKUOPTLCL-UHFFFAOYAA
2: InChI=1/C10H11N/c11-5-8-9-6-1-2-7(4-3-6)10(8)9/h1-2,6-10H,3-4H2
InChIKey=XHTNELKCCFLXEU-UHFFFAOYAJ
3: InChI=1/C10H14/c1-2-8-4-3-7(1)9-5-6-10(8)9/h1-2,7-10H,3-6H2
InChIKey=OYPVZSANECKQOK-UHFFFAOYAR