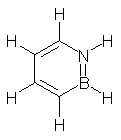
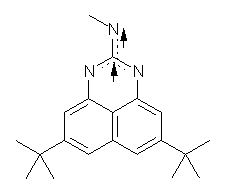
I missed this when it came out, but Quast, Sander and Borden have made the very interesting non-Kekule diradical 1.1
31
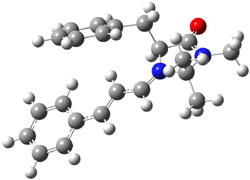
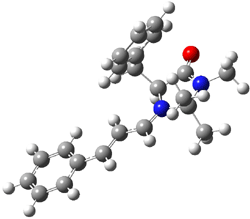
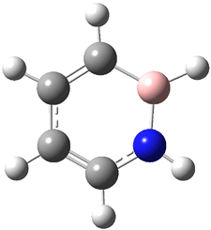
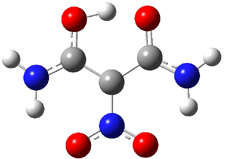
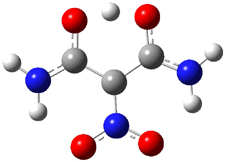
The EPR spectra shows the characteristic six-line signal, with zero-field splitting parameters consistent with related triplet diradicals. The Curie-Weiss plot is linear from 4.6 to 22.9 K. These data suggest a triplet ground state. CASSCF(14,14)/6-31G* computations indicate that the triplet lies 8.5 kcal mol-1 below the singlet. The optimized triplet geometry is shown in Figure 1. The triplet ground state is consistent with the Borden-Davidson rules for radicals.2
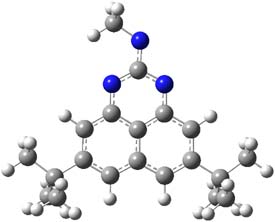 31 |
Figure 1. CASSCF(14,14)/6-31G* optimized structure of triplet 1.
References
(1) Quast, H.; Nudling, W.; Klemm, G.; Kirschfeld, A.; Neuhaus, P.; Sander, W.; Hrovat, D. A.; Borden, W. T., "A Perimidine-Derived Non-Kekule Triplet Diradical," J. Org. Chem. 2008, 73, 4956-4961, DOI: 10.1021/jo800589y.
(2) Borden, W. T.; Davidson, E. R., "Effects of electron repulsion in conjugated hydrocarbon diradicals," J. Am. Chem. Soc. 1977, 99, 4587-4594, DOI: 10.1021/ja00456a010.
InChIs
1: InChI=1/C20H27N3/c1-19(2,3)13-8-12-9-14(20(4,5)6)11-16-17(12)15(10-13)22-18(21-7)23-16/h8-11H,1-7H3,(H2,21,22,23)/f/h22-23H
InChIKey=XAKUHDACNAUAAB-PDJAEHLQCL