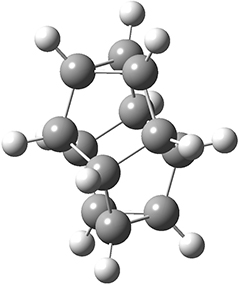
I have been delinquent in writing about the dehydro-Diels-Alder reactions, but really can’t put it off any further. These sets of reactions really deserve a fuller analysis than I am going to summarize here, but this post will provide a good jumping off point for anyone interested in further investigation.
So the Diels-Alder reaction is among the most famous and most important reactions in organic chemistry. The reaction creates a 6-member ring and sets up to four stereocenters. In the past couple of years many chemists have expressed interest in the variant where the four-carbon component is more highly unsaturated, i.e. enyne or diyne. I will summarize the results of three recent computational papers dealing with the reaction of a diyne with an yne.
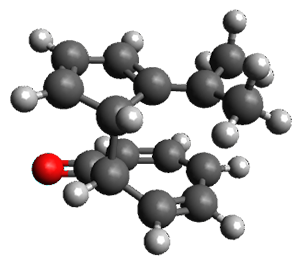
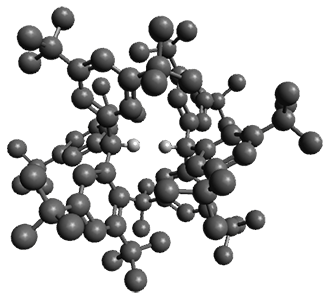
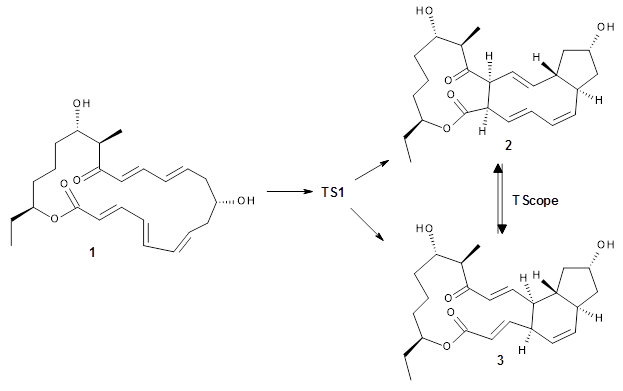
The first paper is by Skraba-Joiner, Johnson, and Agarwal.1 They discuss, among a number of interesting pericyclic reactions, the intramolecular Diels-Alder reaction of triyne 1 to give 2. They examined a concerted and stepwise pathway at (U)M05-2X/6-311+G(d,p) and find the concerted to be favored by 6.0 kcal mol-1. CCSD(T) using these geometries increases the difference to 8.2 kcal mol-1. The T1 diagnostic is fairly large for both the concerted and stepwise transition states, so they also performed CCSD(T)/CBS computations, which had much lower T1 values. The concerted TS remained favorable, but by only 2.7 kcal mol-1.
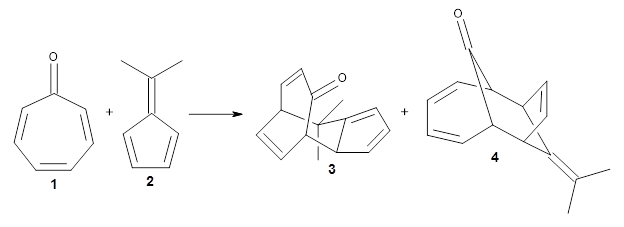
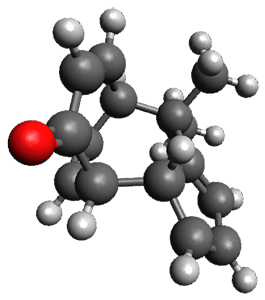
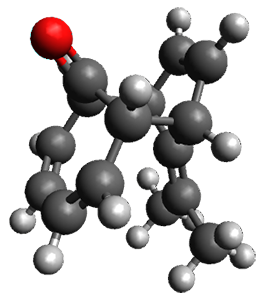
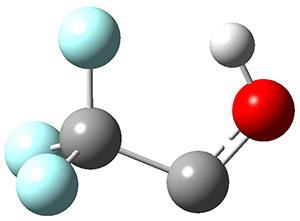
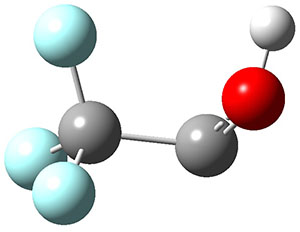
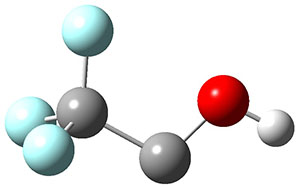
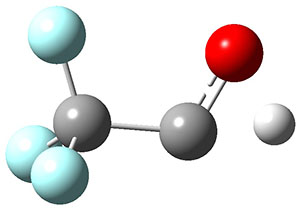
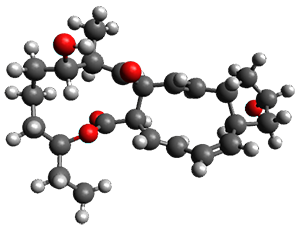
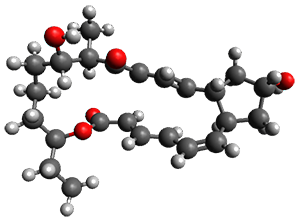
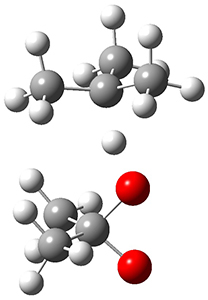
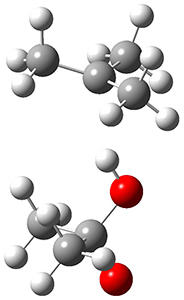
In the same special issue of the Journal of Organic Chemistry, Cramer, Hoye, and Kuwata examined a reaction closely related to what Johnson examined above.2 They looked at the reaction taking 3 into 4 via both experiments and computations. The M06-2x/6-311+G(d,p) geometries for the concerted and first TS along the stepwise path (with R1=R2=H) are shown in Figure 1. Evaluating the energies at SMD(o-dichlorobenzene)/B3LYP-D3BJ/6-311+G-(d,p)//M06-2X/6-311+G(d,p) find in this case (along with all of the other R1/R2 variants they examined) that the stepwise path has a lower barrier than the concerted path. In the case where R1=R2=H, the stepwise path is favored by 6.0 kcal mol-1. Additionally, these stepwise barriers are in reasonable agreement with the experimentally-derived barriers.
Figure 1. M06-2x/6-311+G(d,p) optimized geometries of the concerted and stepwise TSs for the reaction of 3H going to 4H.
It should be pointed out that the wavefunctions for the concerted TSs were all found to be unstable with regard to a restricted to unrestricted relaxation. Given this problem, they also performed a CASPT2 energy evaluation of the concerted and stepwise transition states for the case R1=R2=H. CASPT2 finds the stepwise barrier to be 3.7 kcal mol-1 lower than the concerted barrier.
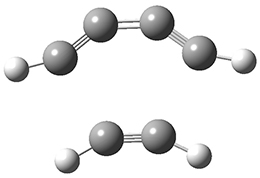
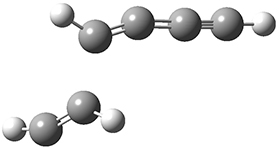
The last paper comes from the Houk lab, and examines the simplest set of intermolecular dehdro-Diels-Alder reactions.3 I will focus here on the most unsaturated analogue, the reaction of 1,3-butadiyne 5 with ethyne to give benzyne 6.
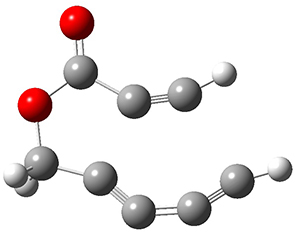
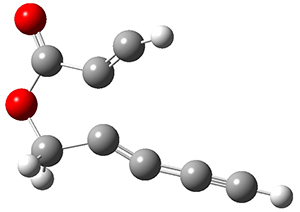
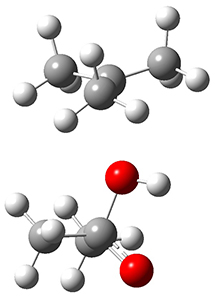
The concreted and stepwise transition states for this reaction (at (U)M06-2X/6-311+G(d,p)) are shown in Figure 2. The concerted barrier is 36.0 kcal moml-1 while the stepwise barrier is slightly lower: 35.2 kcal mol-1. The distortion energy for the concerted reaction is large (43.2 kcal mol-1) due mostly to angle changes in the diyne. Its interaction energy is -7.2 kcal mol-1, similar to the interaction energy in other similar Diels-Alder reactions. In contrast, the distortion energy for the stepwise pathway is 27.5 kcal mol-1, but the interaction energy is +7.7 kcal mol-1. These values are very similar to the distortion and interaction energy of the related (but less saturated DA reactions).
Figure 2. (U)M06-2X/6-311+G(d,p) optimized concerted and stepwise TS for the reaction of 1,3-diyne with ethyne.
Molecular dynamics trajectories for both the concerted and stepwise paths reveal interesting differences. The concerted trajectories show an oscillatory behaviour of bending the angles at the C2 and C3 carbons prior to the TS, and then near synchronous formation of the new C-C bonds. The trajectories initiated at the stepwise TS show no systematic motion. Once the bond is formed, the biradical exhibits a long lifetime, on the order of picoseconds, much longer than the trajectory runs.
These three studies indicate the nature of the dehydro Diels-Alder reaction is very sensitive to reaction conditions, substituents, solvation, and all other manner of effects and will likely prove an area of interest for some time. It should keep a number of computational chemists busy for some time!
References
(1) Skraba-Joiner, S. L.; Johnson, R. P.; Agarwal, J. "Dehydropericyclic Reactions: Symmetry-Controlled Routes to Strained Reactive Intermediates," J. Org. Chem. 2015, 80, 11779-11787, DOI: 10.1021/acs.joc.5b01488.
(2) Marell, D. J.; Furan, L. R.; Woods, B. P.; Lei, X.; Bendelsmith, A. J.; Cramer, C. J.; Hoye, T. R.; Kuwata, K. T. "Mechanism of the Intramolecular Hexadehydro-Diels–Alder Reaction," J. Org. Chem. 2015, 80, 11744-11754, DOI: 10.1021/acs.joc.5b01356.
(3) Yu, P.; Yang, Z.; Liang, Y.; Hong, X.; Li, Y.; Houk, K. N. "Distortion-Controlled Reactivity and Molecular Dynamics of Dehydro-Diels–Alder Reactions," J. Am. Chem. Soc. 2016, 138, 8247-8252, DOI: 10.1021/jacs.6b04113.
InChIs
1: InChI=1S/C9H8/c1-3-5-7-9-8-6-4-2/h1-2H,5,7,9H2
InChIKey=IYZAZSVBWMMSLQ-UHFFFAOYSA-N
2: InChI=1S/C9H8/c1-2-5-9-7-3-6-8(9)4-1/h1,4H,3,6-7H2
InChIKey=PZJMTUKDGZUDBH-UHFFFAOYSA-N
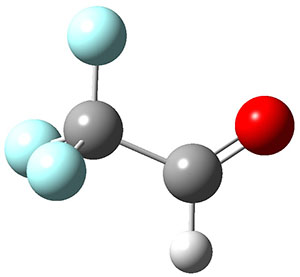
3H: InChI=1S/C8H4O2/c1-3-5-6-7-10-8(9)4-2/h1-2H,7H2
InChIKey=MGXDIFXPYGGQLF-UHFFFAOYSA-N
4H: InChI=1S/C10H8O4/c1-6(11)14-8-2-3-9-7(4-8)5-13-10(9)12/h2-4H,5H2,1H3
InChIKey=GEFLHLNIKGXWCA-UHFFFAOYSA-N
5: InChI=1S/C4H2/c1-3-4-2/h1-2H
InChIKey=LLCSWKVOHICRDD-UHFFFAOYSA-N
6: InChI=1S/C6H4/c1-2-4-6-5-3-1/h1-4H
InChIKey=KLYCPFXDDDMZNQ-UHFFFAOYSA-N