Organic chemists are beginning to recognize that tunneling may be more pervasive than previously thought. This blog has noted a number of interesting occurrences of tunneling, and here’s one more, by Karmakar and Datta.1
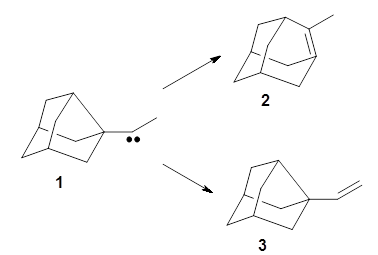
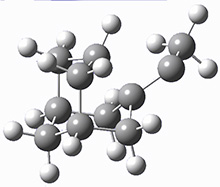
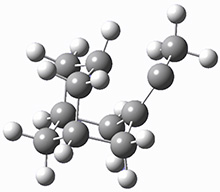
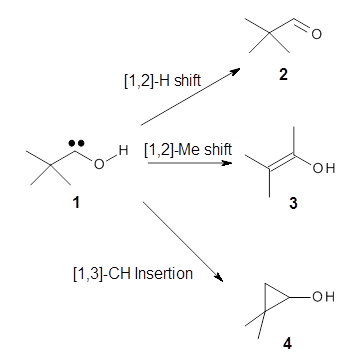
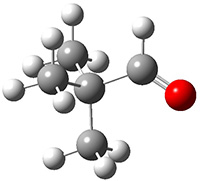
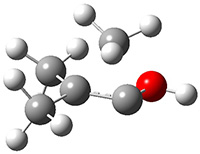
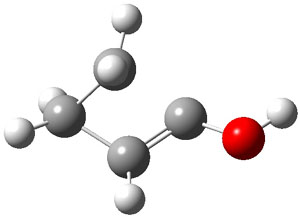
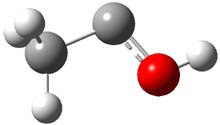
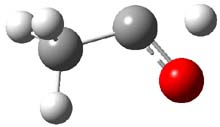
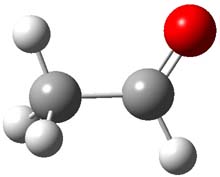
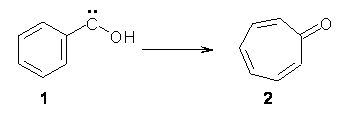
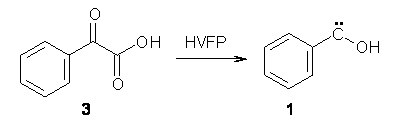
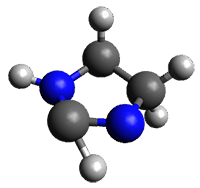
The barrier for the intramolecular earrangement (Reaction 1) taking the carbene 1 into 2 is estimated to be 44.1 kcal mol-1 at M06-2X/6-31+G(d,p), prohibitively large. However, the intermolecular rearrangement (Reaction 2) has a much smaller barrier of 11.4 kcal mol-1. The structures of the transition states for these two reactions are shown in Figure 1.
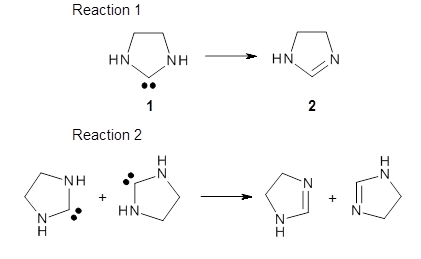
TSintra |
TSinter |
Figure 1. M06-2X/6-31+G(d,p) optimized transition states for Reactions 1 and 2.
Given that the barrier width is likely to be very small for the intramolecular route, perhaps tunneling may play a role. The rate predicted with canonical variational transition-state theory (CVT) and small curvature tunneling (SCT) at 298K is negligible. However, for the intermolecular process, the rate at 298K including tunneling is 3.56 x 104 s-1, more than 10 times great than predicted with CVT alone, and tunneling makes a dramatically larger difference at lower temperatures.
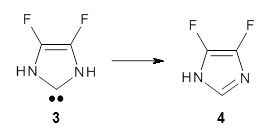
The intermolecular barrier for the rearrangement of 3 into 4 is very small, only 1.6 kcal mol-1.
This manifests in a very interesting rate prediction: the reaction is actually predicted to be slower at temperatures above 150K when tunneling is included than when tunneling is omitted. This is a result of quantum mechanical reflection off of the barrier, and this becomes noticeable with the very small barrier. In addition, the kinetic isotope effects are smaller than expected when D is substituted in for H. These predictions await experimental confirmation.
References
(1) Karmakar, S.; Datta, A. "Tunneling Assists the 1,2-Hydrogen Shift in N-Heterocyclic Carbenes," Angew. Chem. Int. Ed. 2014, 53, 9587-9591, DOI: 10.1002/anie.201404368.
InChIs:
1: InChI=1S/C3H6N2/c1-2-5-3-4-1/h4-5H,1-2H2
InChIKey=JKQUEGZDRZXJNY-UHFFFAOYSA-N
2: InChI=1S/C3H6N2/c1-2-5-3-4-1/h3H,1-2H2,(H,4,5)
InChIKey=MTNDZQHUAFNZQY-UHFFFAOYSA-N
3: InChI=1S/C3H2F2N2/c4-2-3(5)7-1-6-2/h6-7H
InChIKey=LHUPDFSUHVZFPD-UHFFFAOYSA-N
4: InChI=1S/C3H2F2N2/c4-2-3(5)7-1-6-2/h1H,(H,6,7)
InChIKey=KXXZDIFMEWOLPE-UHFFFAOYSA-N