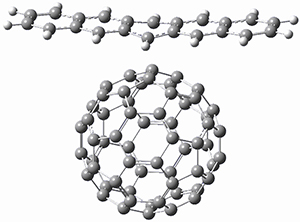
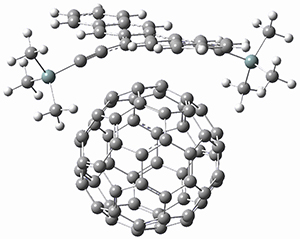
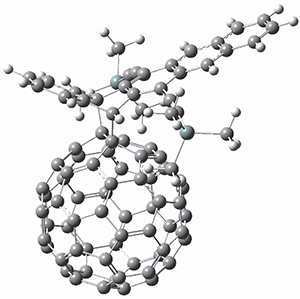
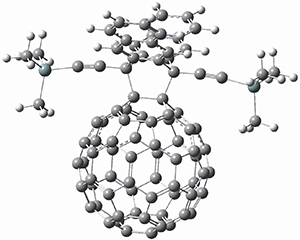
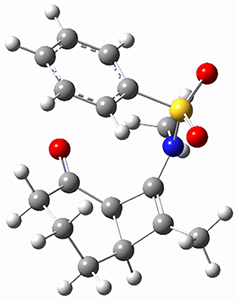
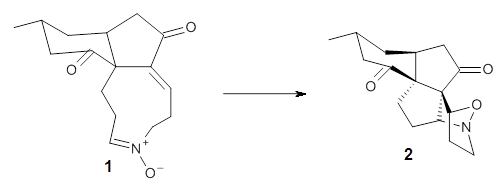
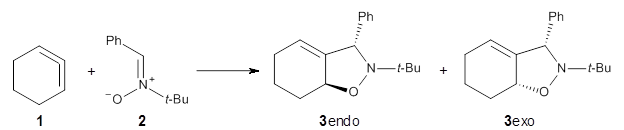
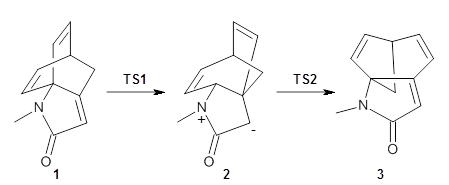1,2-Cyclohexadiene 1 is a very strained and highly reactive species. Houk, Garg and co-workers report on its use as the ene component in a cyclization with a 1,3-dipole, namely nitrones.1 For example, 1 reacts with nitrone 2 to give the cycloadducts 3a and 3b in a ratio of 8.9:1.
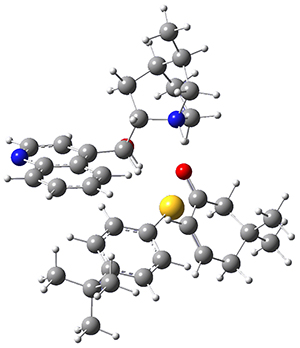
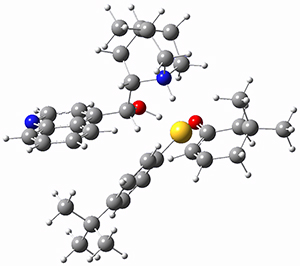
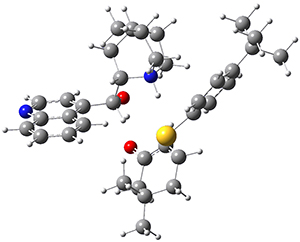
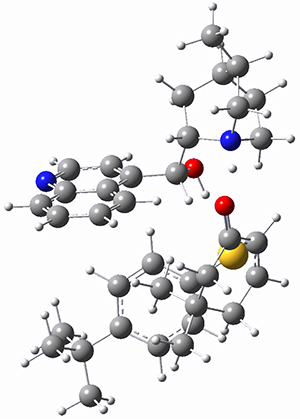
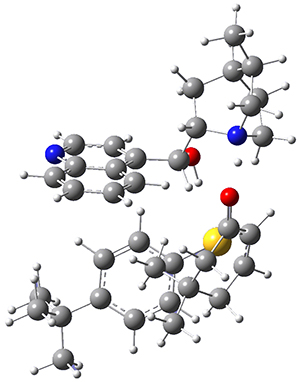
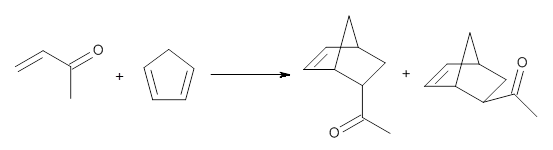
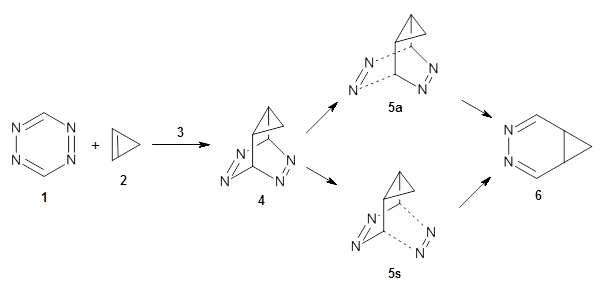
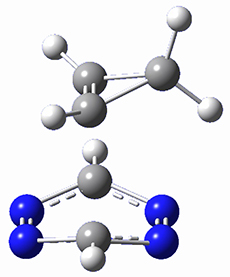
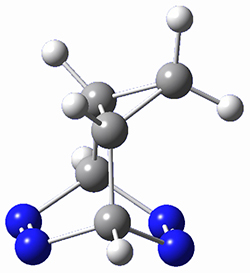
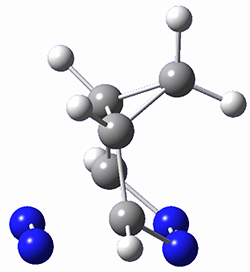
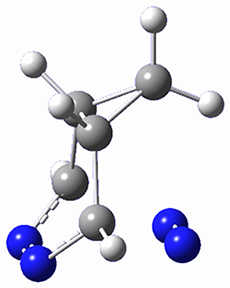
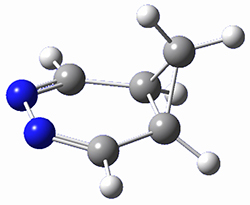
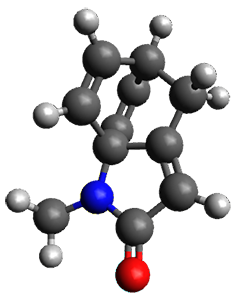
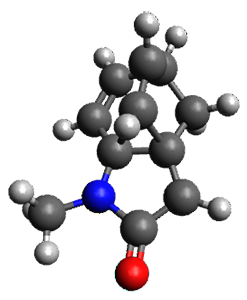
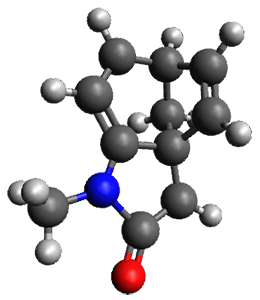
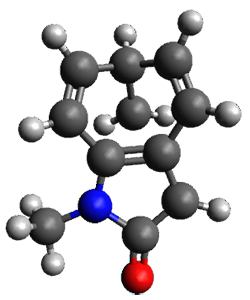
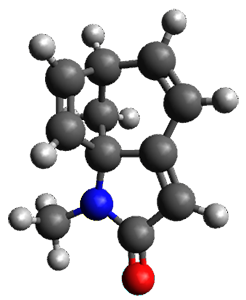
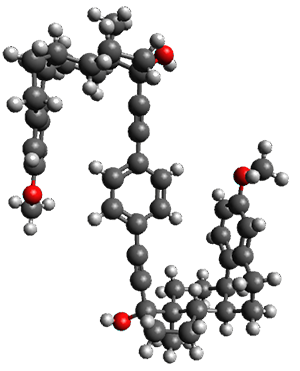
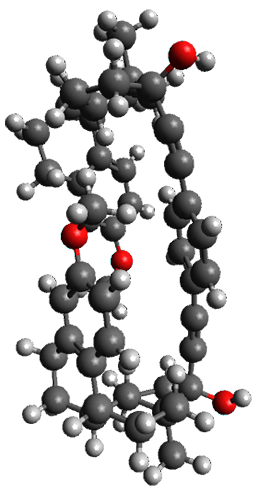
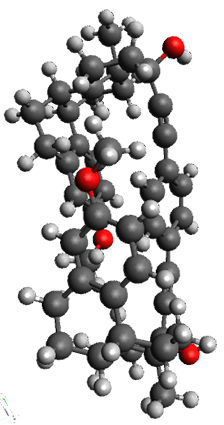
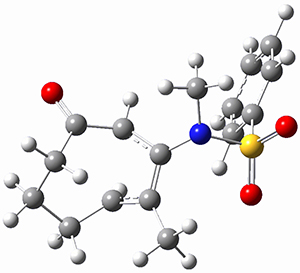
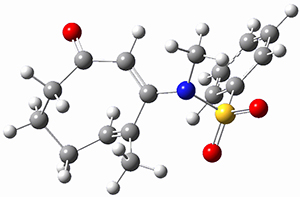
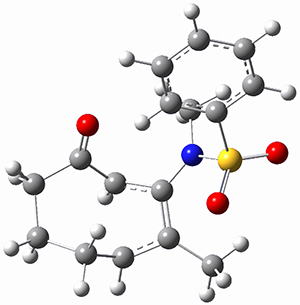
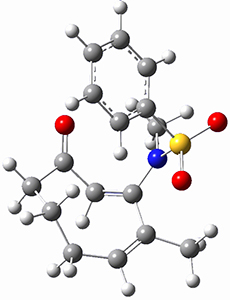
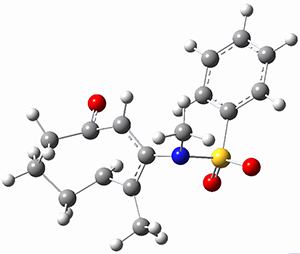
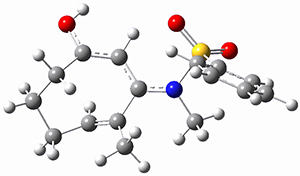
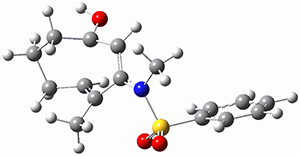
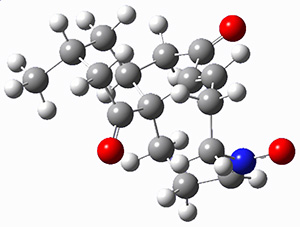
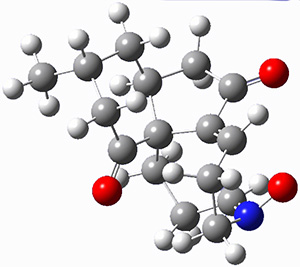
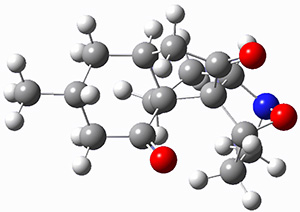
To investigate the mechanism of this reaction, they optimized the structures of all compounds at CPCM(acetonitrile)B3LYP/6-31G(d) and single-point energies were obtained using the B3LYP-D3 functional. The structures of some pertinent critical points are shown in Figure 1. They did locate a concerted transition state (TS1) leading to 3a, with a barrier of 14.5 kcal mol-1, but could not find a concerted TS leading to 3b. (Also, the barriers leading to the other regioisomer are much higher than the ones leading to the observed products.) Rather, they identified a stepwise transition state (TS2) with a barrier of nearly the same energy (14.4 kcal mol-1) that leads to the intermediate (INT), which lies 16.5 kcal mol-1 below reactants. They located two transition states from his intermediate, TS3a and TS3b, leading to the two different products. The barrier to 3a is 1.2 kcal mol-1 lower than the barrier leading to 3b, and this corresponds nicely with the observed diastereoselectivity.
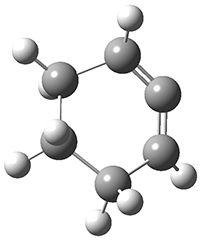
1 |
||
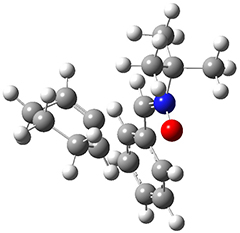
TS1 |
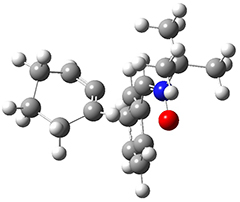
TS2 |
|
|
|
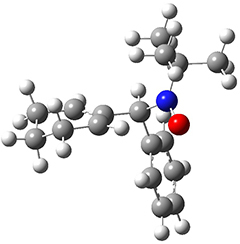
INT |
|
|
|
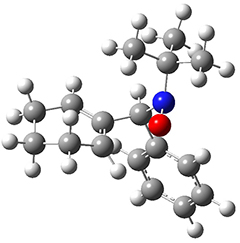
TS3a |
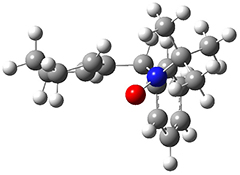
TS3b |
Figure 1. CPCM(acetonitrile)B3LYP/6-31G(d) optimized geometries and CPCM(acetonitrile)B3LYP-D3/6-31G(d) free energies.
References
(1) Barber, J. S.; Styduhar, E. D.; Pham, H. V.; McMahon, T. C.; Houk, K. N.; Garg, N. K.
"Nitrone Cycloadditions of 1,2-Cyclohexadiene," J. Am. Chem. Soc. 2016, 138, 2512-2515, DOI: 10.1021/jacs.5b13304.
InChIs
1: InChI=1S/C6H8/c1-2-4-6-5-3-1/h1,5H,2,4,6H2
InChIKey=NMGSDTSOSIPXTN-UHFFFAOYSA-N
2: InChI=1S/C11H15NO/c1-11(2,3)12(13)9-10-7-5-4-6-8-10/h4-9H,1-3H3/b12-9-
InChIKey=IYSYLWYGCWTJSG-XFXZXTDPSA-N
3a: InChI=1S/C17H23NO/c1-17(2,3)18-16(13-9-5-4-6-10-13)14-11-7-8-12-15(14)19-18/h4-6,9-11,15-16H,7-8,12H2,1-3H3/t15-,16-/m0/s1
InChIKey=HQIBSHJEOJPFTI-HOTGVXAUSA-N
3b: InChI=1S/C17H23NO/c1-17(2,3)18-16(13-9-5-4-6-10-13)14-11-7-8-12-15(14)19-18/h4-6,9-11,15-16H,7-8,12H2,1-3H3/t15-,16+/m1/s1
InChIkey=HQIBSHJEOJPFTI-CVEARBPZSA-N