At the Spring ACS meeting in New Orleans this past April, Jonathan Goodman told me about his group’s new work on computed NMR spectra. This study has now appeared.1 The new aspect of this work is taking on compounds with significant conformational flexibility. They first examined the bifuranyl- and pyranopyrans acetals 1-6.
|
|
|
|
|
|
|
|
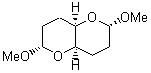
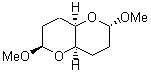
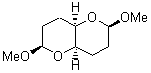
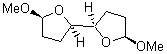
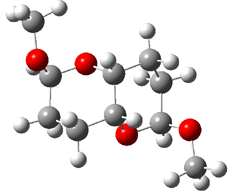
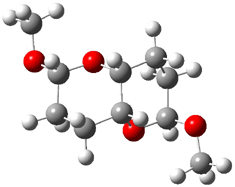
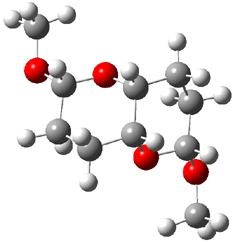
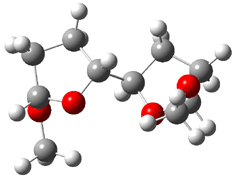
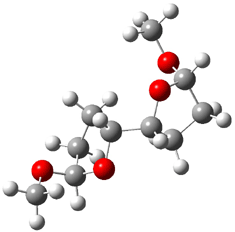
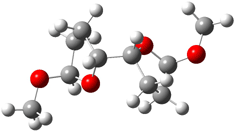
They identified all energetically low-lying conformers through a Monte Carlo MM search, and reoptimized the structures at B3LYP/6-31G**. PCM energies, with the dielectric set to 4.81 to simulate CHCl3, were obtained using the gas-phase geometries. 13C NMR shifts were then computed and weighted based on Boltzmann averaging. Compounds 4-7 require consideration of 4, 5, or 7 conformations, respectively, to account for at least 95% of the population. (The lowest energy conformation for each compound is displayed in Figure 1.) The computed 13C NMR chemical shifts were then compared against the experimental values that have been obtained for 5 of these 6 compounds, though it was not known which experimental spectra corresponds with which structure. Based on the correlation coefficients and the mean and maximum values of the differences between the calculated and experimental shifts allows for identification of the structure that corresponds to each of the five spectra.
 1 |
 2 |
 3 |
 4 |
 5 |
 6 |
Figure 1. B3LYP/6-31G** minimum energy conformation of 1-6.1
They next took on the absolute structure of elatenyne 7. This compound has been isolated and its 13C NMR obtained nd interpreted.2 The compound can exist in one of 32 different diastereomers. Instead of having to stereospecifically synthesize each diastereomer, obtain its NMR spectra and compare with the natural product, Goodman suggests that one can compute the 13C NMR spectra for each isomer, identified the likely candidates, and synthesize just those if verification is necessary. So, they computed the spectra of all 32 isomers following the above prescription. The chemical shifts for the carbons bearing bromine have relatively large errors, though these can by systematically corrected by reducing the computed values by about 20 ppm. Comparison of the computed spectra of each isomer with the experimental spectra gave three possible isomers (7a-c, see Figure 2) with very strong correlation coefficients. Examination of the men value of the difference of the computed chemical shifts with the experimental values (not including the carbon atoms with a bromine attached) suggests one more possibility 7d (see Figure 2).
|
|
|
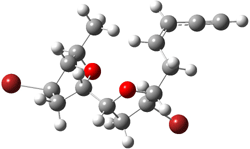 7a |
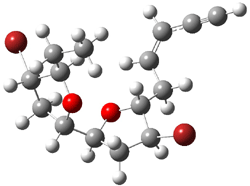 7b |
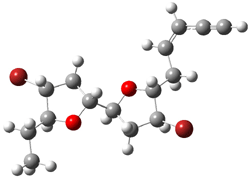 7c |
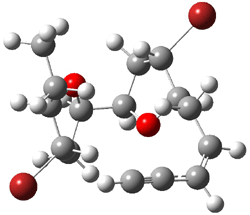 7d |
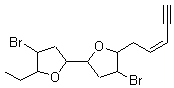
Figure 2. B3LYP/6-31G** minimum energy conformation of 7a-d.1
References
(1) Smith, S. G.; Paton, R. S.; Burton, J. W.; Goodman, J. M., "Stereostructure Assignment
of Flexible Five-Membered Rings by GIAO 13C NMR Calculations: Prediction of the Stereochemistry of Elatenyne," J. Org. Chem., 2008, DOI: 10.1021/jo8003138.
(2) Sheldrake, H. M.; Jamieson, C.; Burton, J. W., “The Changing Faces of Halogenated Marine Natural Products: Total Synthesis of the Reported Structures of Elatenyne and an Enyne from Laurencia majuscula,” Angew. Chem. Int. Ed., 2006, 45, 7199-7202, DOI: 10.1002/anie.200602211
InChIs
1-3: InChI=1/C10H18O4/c1-11-9-5-3-8-7(13-9)4-6-10(12-2)14-8/h7-10H,3-6H2,1-2H3
4-6: InChI=1/C10H18O4/c1-11-9-5-3-7(13-9)8-4-6-10(12-2)14-8/h7-10H,3-6H2,1-2H3
7: InChI=1/C15H20Br2O2/c1-3-5-6-7-13-11(17)9-15(19-13)14-8-10(16)12(4-2)18-14/h1,5-6,10-15H,4,7-9H2,2H3/b6-5-
InChIKey=SKSTYQSRJPCZSW-WAYWQWQTBQ

Jonathan Goodman responded on 28 May 2008 at 10:01 am #
Thank you for highlighting our paper. There are two conclusions that come out of this study, which we think are of general interest:
(i) Use MMFF conformation searches and then B3LYP/6-31G** single point calculations
We expected we would have to remininise all the conformations at the DFT level. We found that single point DFT calculations on the MMFF geometries gave results of very similar accuracy, and were an order of magnitude faster.
(ii) Omit carbons with halogens attached
We experimented with various ways of correcting for the large errors in the C-Br signals. Including a 20 ppm correction works quite well, but our preferred approach is simply to omit these signals from the analysis. This gave us similar clarity, without introducing a rather arbitrary correction. The stereochemistry at bromine is important in the molecules under study, but it has an effect on the neighbouring stereocentres which relay the chiral information. After omitting these difficult carbon atoms, we could still distinguish the diastereoisomers effectively.