The weak n→π* interaction has been proposed to explain some conformational structure. Singh, Mishra, Sharma, and Das have now provided the first spectroscopic evidence of this interactions.1 They examined the structure of phenylformate 1. This compound can exist as two conformational isomers, having the carbonyl oxygen pointing towards (cis) or away (trans) from the phenyl ring. They optimized the structures of these two conformers at M05-2X/aug-cc-pVDZ and find that the cis isomer is lower in energy by 1.32 kcal mol-1. Unfortunately, the authors do not provide the structures of these isomers, but since they are so small, I reoptimized them at ωB97XD/6-311g(d) and they are displayed in Figure 1. At this computational level, the cis isomer is lower in enthalpy than the trans isomer by 1.35 kcal mol-1.
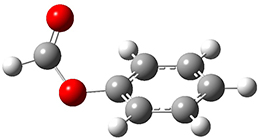
1cis |
1trans |
Figure 1. ωB97XD/6-311g(d) optimized structures of the cis and trans conformations of 1.
One-color resonant 2-photon ionization (1C-R2PI) spectroscopy followed by UV-VIS hole burning spectroscopy identified two isomers of 1, one present in greater amount that the other. The IR spectra of the dominant isomer showed a carbonyl stretch at 1766 cm-1, in nice agreement with the predicted frequency of 1cis (1770 cm-1). The carbonyl stretch for the minor isomer is at 1797 cm-1, again in nice agreement with the computed frequency for 1trans (1800 cm-1). The cis isomer has the lower carbonyl frequency due to partial donation of the carbonyl oxygen electrons to the π* orbital of the phenyl ring.
References
(1) Singh, S. K.; Mishra, K. K.; Sharma, N.; Das, A. "Direct Spectroscopic Evidence for an n→π* Interaction," Angew. Chem. Int. Ed. 2016, 55, 7801-7805, DOI: 10.1002/anie.201511925.
InChIs
1: InChI=1S/C7H6O2/c8-6-9-7-4-2-1-3-5-7/h1-6H
InChIKey=GEOWCLRLLWTHDN-UHFFFAOYSA-N

Steven Bachrach responded on 20 Jul 2016 at 12:59 pm #
Thanks to Alan Shusterman for pointing out that compound 1 is phenylformate not methylformate. This corrections has been made to the post.