Here’s a real tour de force study combining exciting experiments with detailed computations. It’s a look at the radical cation of propellane performed by Bally and Williams.1 This paper has been nicely reviewed by Hiberty.2
Propellane 1, whose bridgehead-bridgehead bond has been a topic of an earlier post, has a HOMO that is largely outside of the bridgehead-bridgehead region. Thus, loss of an electron to form the radical cation 1.+ seems unlikely to lead to any significant geometrical change. However, the ESR of the radical cation of propellane shows two types of hydrogens, one type of four hydrogens and a second type of two hydrogens. This is incompatible with a D3h structure similar to that of 1. Furthermore, loss of an electron from dimethylenecyclopropane 2 leads to a species whose ESR is nearly identical to that of the radical cation of propellane. Analysis of the ESR suggests that the radical actually produced is 3.+.
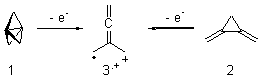
CCSD(T)/cc-pVTZ//B3LYP/6-31G* computations were performed to try to discern a mechanism for this rearrangement. The D3h structure of 1.+ is a local energy minimum with most computational methods, though not with B3LYP, where it is a TS connecting mirror image C2 structures. Breaking symmetry to C2 leads to a TS (TS1) for cleaving one of the C-Cbridgehead bonds. This TS is only 1.15 kcal mol-1 above 1.+, and leads to 4.+, 7.38 kcal mol-1 below 1.+. Cleavage of a second C-Cbridgehead bond passes through TS2, with a barrier from 4.+ of only 2.89 kcal mol-1. This leads to 2.+. Lastly, cleavage of a third C-Cbridgehead bond through TS3, with a barrier of only 2.09 kcal mol-1 above 2.+, leads to 3.+, overall 30.4 kcal mol-1 exothermic from 1.+. The structures of these critical points are shown in Figure 1. Quite a neat little pathway – three sequential bond ruptures without ever cleaving what was the weakest bond in the original compound (the bridgehead-bridgehead bond)!
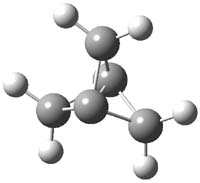 1.+ |
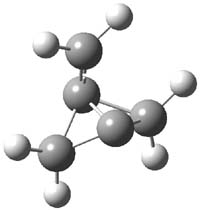 TS1 |
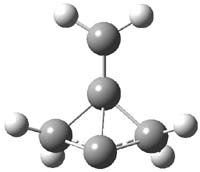 4.+ |
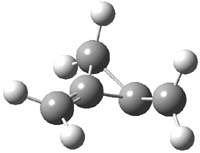 TS2 |
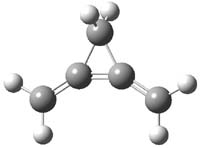 2.+ |
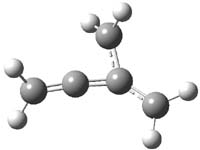 TS3 |
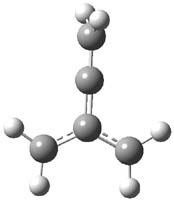 3.+ |
|
Table 1. B3LYP/6-31G* optimized critical points on the pathway of 1.+ to 3.+.
Relative energies in kcal mol-1
The cool part of this is why the barrier is so small leading out of 1.+ – vibronic coupling via Cs distortion of 1.+ with its first excited state leads to an energy lowering of this pathway. This sort of vibronic coupling had in fact been implicated by Heilbronner and Wiberg3 in arguing the photoelectron spectrum of 1.
References
(1) Müller, B.; Bally, T.; Pappas, R.; Williams, F., "Spectroscopic and Computational Studies on the Rearrangement of Ionized [1.1.1]Propellane and Some of its Valence Isomers: The Key Role of Vibronic Coupling," J. Am. Chem. Soc. 2010, 132, 14649-14660, DOI: 10.1021/ja106024y
(2) Hiberty, P. C., "Vibronic coupling: Cage-breaking cascade," Nat. Chem. 2011, 3, 96-97, DOI: 10.1038/nchem.971
(3) Honegger, E.; Huber, H.; Heilbronner, E.; Dailey, W. P.; Wiberg, K. B., "The PE spectrum of [1.1.1]propellane: evidence for a non-bonding MO?," J. Am. Chem. Soc., 1985, 107, 7172-7174, DOI: 10.1021/ja00310a068
InChI
1: InChI=1/C5H6/c1-4-2-5(1,4)3-4/h1-3H2
InChIKey=ZTXSPLGEGCABFL-UHFFFAOYAJ
2: InChI=1/C5H6/c1-4-3-5(4)2/h1-3H2
InChIKey=ZNKWTJLYBOAVHI-UHFFFAOYAT
3.+: InChI=1/C5H6/c1-4-5(2)3/h1-3H2/q+1
InChIKey=BVWPXIKADZQKEJ-UHFFFAOYAU
